Topics: Biology, COVID-19, Genetics, Research
Abstract
A recent genetic association study1 identified a gene cluster on chromosome 3 as a risk locus for respiratory failure after infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A separate study (COVID-19 Host Genetics Initiative)2 comprising 3,199 hospitalized patients with coronavirus disease 2019 (COVID-19) and control individuals showed that this cluster is the major genetic risk factor for severe symptoms after SARS-CoV-2 infection and hospitalization. Here we show that the risk is conferred by a genomic segment of around 50 kilobases in size that is inherited from Neanderthals and is carried by around 50% of people in south Asia and around 16% of people in Europe.
Main
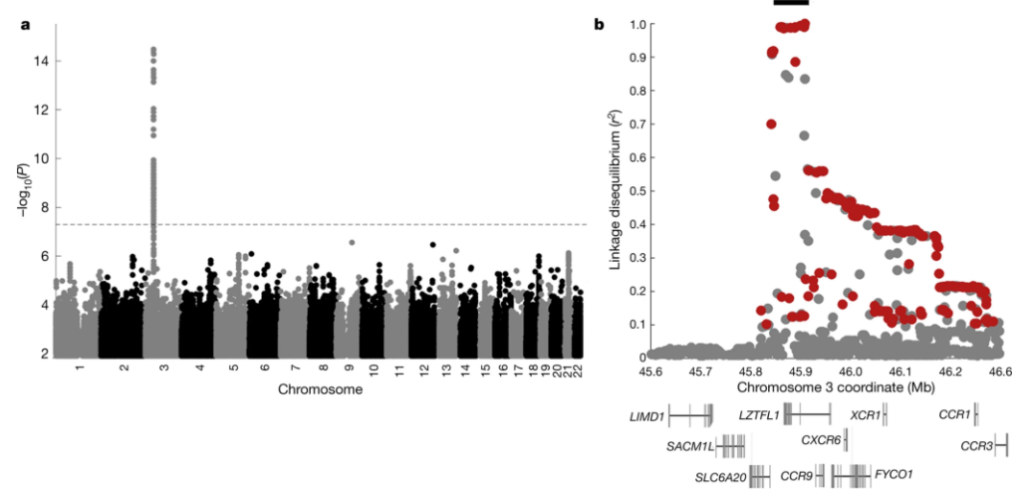
The COVID-19 pandemic has caused considerable morbidity and mortality, and has resulted in the death of over a million people to date3. The clinical manifestations of the disease caused by the virus, SARS-CoV-2, vary widely in severity, ranging from no or mild symptoms to rapid progression to respiratory failure4. Early in the pandemic, it became clear that advanced age is a major risk factor, as well as being male and some co-morbidities5. These risk factors, however, do not fully explain why some people have no or mild symptoms whereas others have severe symptoms. Thus, genetic risk factors may have a role in disease progression. A previous study1 identified two genomic regions that are associated with severe COVID-19: one region on chromosome 3, which contains six genes, and one region on chromosome 9 that determines ABO blood groups. Recently, a dataset was released by the COVID-19 Host Genetics Initiative in which the region on chromosome 3 is the only region that is significantly associated with severe COVID-19 at the genome-wide level (Fig. 1a). The risk variant in this region confers an odds ratio for requiring hospitalization of 1.6 (95% confidence interval, 1.42–1.79) (Extended Data Fig. 1).
The genetic variants that are most associated with severe COVID-19 on chromosome 3 (45,859,651–45,909,024 (hg19)) are all in high linkage disequilibrium (LD)—that is, they are all strongly associated with each other in the population (r2 > 0.98)—and span 49.4 thousand bases (kb) (Fig. 1b). This ‘core’ haplotype is furthermore in weaker linkage disequilibrium with longer haplotypes of up to 333.8 kb (r2 > 0.32) (Extended Data Fig. 2). Some such long haplotypes have entered the human population by gene flow from Neanderthals or Denisovans, extinct hominins that contributed genetic variants to the ancestors of present-day humans around 40,000–60,000 years ago6,7. We therefore investigated whether the haplotype may have come from Neanderthals or Denisovans.
The major genetic risk factor for severe COVID-19 is inherited from Neanderthals, Hugo Zeberg, & Svante Pääbo, Nature
